Methods of Phylogenetic Analysis
Understanding evolutionary relationships requires accurate analytical tools. Therefore, scientists use structured tree-building approaches to study species history. This article of Methods of Phylogenetic Analysis explains three major methods: Maximum Parsimony (MP), Maximum Likelihood (ML), and Bayesian Inference (BI). Although all three reconstruct evolutionary trees, they differ in logic, statistical framework, and computational demand. You can easily download this note as a PDF using the link provided just below the post for quick access and offline reading.
Systematics Notes | Systematics PPTs | Evolution Notes | Evolution PPTs
Definition
Phylogenetic Methods Comparison refers to the systematic evaluation of Maximum Parsimony, Maximum Likelihood, and Bayesian Inference methods used to reconstruct evolutionary trees from molecular or morphological data using simplicity, likelihood estimation, or probability-based reasoning.
Phylogenetic Methods Comparison: Fundamental Principles
Each method follows a different scientific philosophy. Therefore, understanding their core ideas is essential before selecting a tree-building approach.
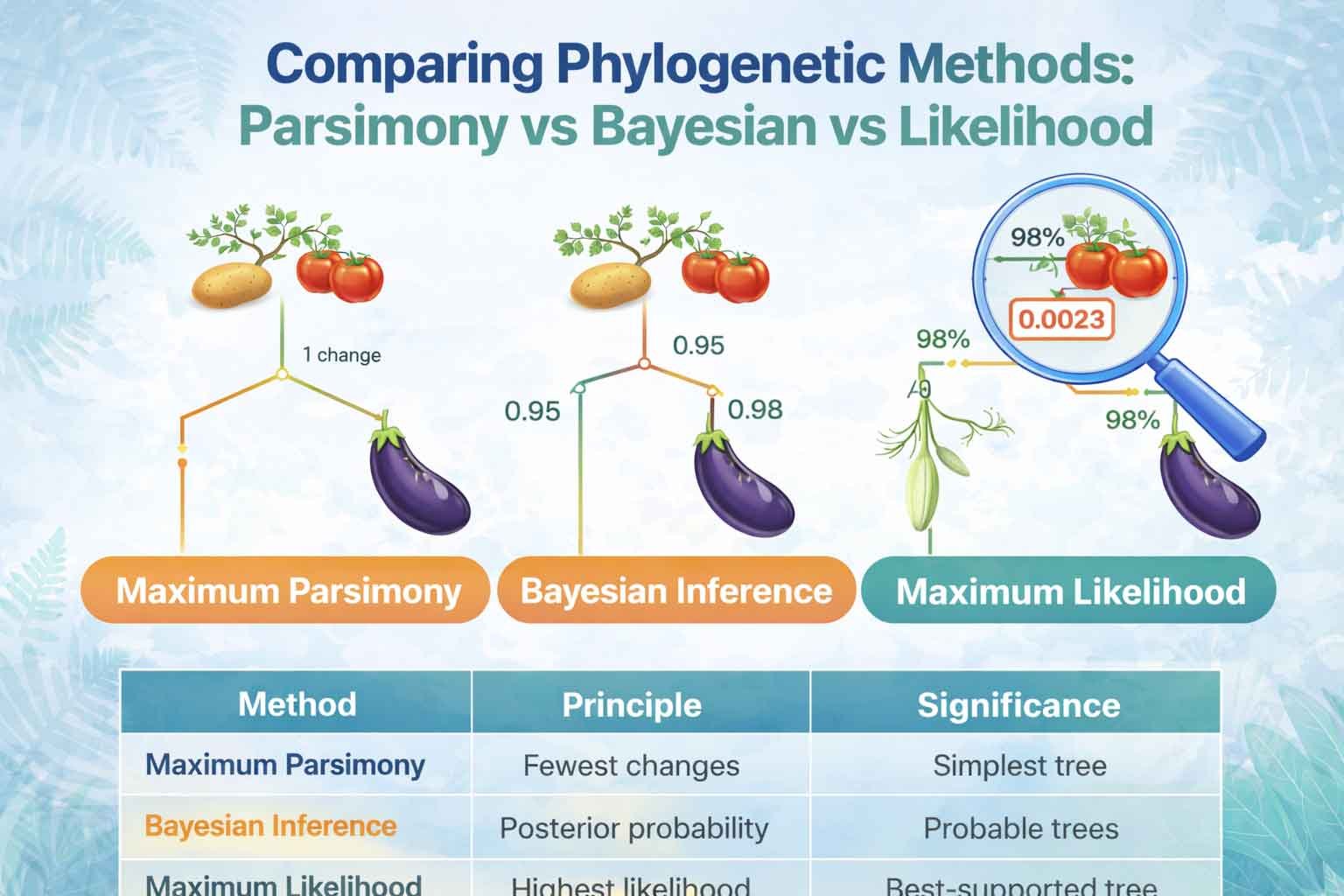
1. Maximum Parsimony (MP)
Basic Principle
Maximum Parsimony is based on simplicity. It selects the evolutionary tree that requires the fewest character changes. In other words, it assumes evolution follows the shortest path.
Methodology
- Aligning DNA, RNA, or protein sequences
- Generating alternative tree topologies
- Counting character state changes
- Selecting the tree with minimum total changes
Significance
Maximum Parsimony is easy to understand. Moreover, it does not require an explicit substitution model. Therefore, it is useful for morphological datasets and small molecular studies.
Applications
- Morphological character analysis
- Early molecular phylogenetics
- Preliminary tree construction
Limitations
- Sensitive to homoplasy
- Less reliable when evolutionary rates vary
- Performs poorly with large datasets
You may also like NOTES in... BOTANY BIOCHEMISTRY MOL. BIOLOGY ZOOLOGY MICROBIOLOGY BIOSTATISTICS ECOLOGY IMMUNOLOGY BIOTECHNOLOGY GENETICS EMBRYOLOGY PHYSIOLOGY EVOLUTION BIOPHYSICS BIOINFORMATICS
2. Maximum Likelihood (ML)
Basic Principle
Maximum Likelihood selects the tree with the highest probability of generating the observed data under a chosen evolutionary model. Thus, it evaluates statistical fit rather than simplicity.
Methodology
- Selecting a substitution model
- Generating possible tree topologies
- Calculating likelihood scores
- Choosing the tree with the highest likelihood value
Significance
Maximum Likelihood is statistically robust. In addition, it handles variable evolutionary rates effectively. Therefore, it is widely used in modern molecular systematics.
Applications
- Molecular phylogenetic analysis
- Evolutionary rate estimation
- Comparative genomics
Limitations
- Computational demand is high
- Model selection affects accuracy
- Analysis may be slow for very large datasets
3. Bayesian Inference (BI)
Basic Principle
Bayesian Inference estimates the probability of a tree given the observed data. It combines prior knowledge with likelihood calculations. Thus, it produces posterior probability values for clades.
Methodology
- Selecting an evolutionary model
- Assigning prior probabilities
- Running Markov Chain Monte Carlo (MCMC) simulations
- Estimating posterior probability distributions
Significance
Bayesian Inference directly provides probability support values. Moreover, it allows integration of prior biological knowledge. Therefore, it is powerful for complex evolutionary analyses.
Applications
- Divergence time estimation
- Phylogeographic studies
- Hypothesis testing in evolution
Limitations
- Prior assumptions must be chosen carefully
- Computational cost is high
- Interpretation can be challenging for beginners
Comparative Table of the Three Methods
| Feature | Maximum Parsimony | Maximum Likelihood | Bayesian Inference |
|---|---|---|---|
| Core Idea | Fewest evolutionary changes | Highest likelihood of observed data | Highest posterior probability |
| Statistical Model | No explicit model required | Requires substitution model | Requires model and prior probabilities |
| Computational Demand | Low to moderate | High | High |
| Support Values | Bootstrap values | Bootstrap values | Posterior probabilities |
| Best For | Small datasets, morphology | Large molecular datasets | Complex evolutionary analyses |
| Main Weakness | Sensitive to homoplasy | Computationally intensive | Sensitive to prior assumptions |
Conclusion
This Phylogenetic Methods Comparison shows that no single method is universally superior. The best approach depends on dataset size, data type, research objective, and computational resources. Therefore, selecting the appropriate method ensures accurate reconstruction of evolutionary history.
<<< Back to Systematics Notes Page
You may also like... NOTES QUESTION BANK COMPETITIVE EXAMS. PPTs UNIVERSITY EXAMS DIFFERENCE BETWEEN.. MCQs PLUS ONE BIOLOGY NEWS & JOBS MOCK TESTS PLUS TWO BIOLOGY PRACTICAL
Study Offline!! Download this Notes as a PDF
🌿 Dear Readers,
I hope you found this article helpful and easy to understand. If you have any questions, suggestions, or thoughts, I would truly love to hear from you.
Please share your feedback in the comments below. Your participation helps make EasyBiologyClass a better learning space for everyone.
Best regards,
EasyBiologyClass